
ISO 14971 is the international standard established by the International Organization for Standardization (ISO) for managing risk associated with medical devices, including in vitro diagnostic (IVD) devices and software as a medical device (SaMD). ISO 14971 provides a structured methodology for identifying hazards, analyzing and evaluating risks, implementing control measures, and continuously monitoring and updating risk throughout a medical device’s entire lifecycle.
Risk, as defined by ISO 14971, is the combination of the probability of harm occurring and the severity of that harm. Key concepts include hazards (potential sources of harm), hazardous situations (conditions involving exposure to hazards), and harm itself (injury or damage to health, property, or the environment).
The most current version, ISO 14971:2019, is widely accepted by global regulatory bodies and complements ISO/TR 24971:2020, which provides detailed guidance on implementing ISO 14971 requirements.
The ISO 14971 standard outlines a six-step risk management process: risk analysis, risk evaluation, risk control, evaluation of overall residual risk, risk management review, and production and post-production monitoring. Common tools used to support these steps include failure modes and effects analysis (FMEA), fault tree analysis (FTA), preliminary hazard analysis (PHA), and hazard and operability study (HAZOP).
Effective implementation of ISO 14971 has benefits such as structured risk mitigation, risk control over the product lifecycle, and support for benefit-risk analysis, while also improving audit readiness through comprehensive, up-to-date risk documentation. Although ISO 14971 is not legally mandated, its adoption is often expected by regulators and is critical for ensuring regulatory compliance, enhancing patient safety, and reducing the likelihood of recalls and post-market failures.
To streamline ISO 14971 implementation, companies may opt to use quality management software with risk management capabilities. Quality management software enhances ISO 14971 compliance by automating workflows, centralizing the document repository, and streamlining all risk-related activities and other quality processes.
SimplerQMS is a cloud-based electronic quality management system (eQMS) software built specifically for the medical device and life science industries. SimplerQMS helps organizations meet ISO 14971 and ISO 13485 requirements by offering tools for centralized risk documentation, pre-built templates, automated workflows, and more.
What Is ISO 14971?
ISO 14971 is the international standard for medical device risk management, developed by the International Organization for Standardization (ISO). ISO 14971 outlines a structured process for identifying hazards, analyzing and evaluating risks, applying appropriate controls, and monitoring those controls throughout the entire lifecycle of a medical device.
The core objective of the ISO 14971 standard is to ensure that medical devices are safe for patients, users, and the environment. To achieve this, the ISO 14971 standard provides a structured approach to identifying, analyzing, evaluating, and controlling risks associated with devices, such as electrical failures, biocompatibility concerns, radiation exposure, software defects, and usability issues.
ISO 14971 applies to medical devices, in vitro diagnostic (IVD) devices, and software as a medical device (SaMD). ISO 14971 is accepted by regulatory bodies such as the U.S. Foodand Drug Administration (FDA), European Medicines Agency (EMA), Health Canada, Therapeutic Goods Administration (Australia), and Japan’s Pharmaceuticals and Medical Devices Agency (PMDA).
The current version, ISO 14971:2019, has been updated from the 2007 edition and aligns with the EU MDR/IVDR through Amendment A11:2021 of EN ISO 14971:2019. Additionally, ISO/TR 24971:2020 (Medical devices – Guidance on the application of ISO 14971) provides practical guidance on implementing ISO 14971.
What Is Risk Management?
Risk management, as defined by ISO 14971, is the systematic application of policies, procedures, and practices to analyze, evaluate, control, and monitor risks associated with medical devices across their entire lifecycle. Risk management ensures that devices remain safe, effective, and compliant with global regulatory requirements.
Risk Management Terminology
The following key risk management terms are essential for understanding and implementing effective medical device risk management.
- Hazard: A hazard refers to a potential source of harm, such as an electrical current or a biological agent (pathogen).
- Hazardous Situation: A hazardous situation describes the specific conditions under which individuals, property, or the environment are exposed to that hazard (e.g., a patient touches exposed wiring on a damaged power cord or a patient is exposed to an inadequately sterilized device during surgery).
- Harm: Harm is the actual consequence, such as physical injury or health deterioration, or environmental and property damage (e.g., electrical shock to patient or surgical site infection/sepsis).
- Risk: Risk is the combination of the probability of occurrence of harm and the severity of that harm.
- Residual Risk: Residual risk is the risk that remains after risk control measures have been applied.
- Overall Residual Risk: The overall residual risk is the combined residual risk from all identified hazards, considering all implemented control measures. It represents the total remaining risk associated with the medical device.
- Benefit-risk Analysis: Benefit-risk analysis is a systematic evaluation of whether the clinical benefits of a medical device outweigh its residual risks, especially when those risks cannot be further reduced.
According to Annex B.2 of ISO 14971, the standard defines a structured six-step risk management process for medical devices. The risk management process involves analyzing risks, evaluating their acceptability, applying controls to mitigate them, and assessing the overall residual risk. The process is performed iteratively, ensuring risk activities are fully integrated with QMS processes, including the effective monitoring of post-production data to identify and address emerging risks.
Risk Management Tools and Techniques
A variety of risk analysis tools and techniques can be used to support the identification, evaluation, and control of hazards.
ISO 14971 emphasizes hazard analysis, focusing on situations that could lead to harm, regardless of whether a failure has occurred. However, failure analysis tools, such as FMEA and FTA, can be used as complementary tools to identify potential failure modes affecting device performance, which serve as inputs into the overall hazard analysis. By integrating both hazard-based and failure-based techniques, organizations can achieve a more comprehensive understanding of risk, enhancing the safety and effectiveness of their medical devices.
The common tools and techniques used in risk management include the following.
- Preliminary Hazard Analysis (PHA): PHA supports early product development by identifying and classifying hazards when design details are limited, but risk foresight is crucial for informed planning.
- Failure Modes and Effects Analysis (FMEA): FMEA is used to identify potential failure modes in a system or component, assess their effects, and prioritize them based on severity, occurrence, and detectability.
- Fault Tree Analysis (FTA): FTA visually maps failure logic from an undesired top event down to its root causes, aiding in understanding complex failure relationships and designing mitigation strategies.
- Hazard and Operability Study (HAZOP): HAZOP systematically examines process deviations and their safety impacts on medical devices.
Why Is ISO 14971 Important?
ISO 14971 is important for the medical device industry because it mandates a structured risk management process that enhances patient safety and product reliability by identifying, evaluating, and mitigating potential hazards. Regulatory bodies, such as the FDA and EMA, do not mandate the use of ISO 14971; however, they require comprehensive risk management, making the use of ISO 14971 highly recommended and often expected in submissions.
Moreover, ISO 14971 enables effective and proactive risk management through continuous assessment, traceability, and the integration of feedback. Compliance with ISO 14971 ensures product quality and safety, strengthens market trust, accelerates approvals, and prevents recalls, while non-compliance risks legal consequences, market exclusion, and patient harm.
What Are the Benefits of Implementing ISO 14971?
Implementing ISO 14971 offers the following benefits.
- Enhance Patient Safety: ISO 14971 enhances patient protection by enabling systematic hazard identification, risk assessment, an effective risk control process, and management of residual risk throughout the product lifecycle.
- Achieve Regulatory Compliance: ISO 14971 aligns with FDA 21 CFR 820, EU MDR, and EU IVDR, making it a key standard for demonstrating effective medical device risk management in compliance submissions.
- Maintain Risk Control: ISO 14971 applies a continuous risk management process throughout the device lifecycle, from design to decommissioning, involving ongoing collection and review of production and post-production information to identify new hazards and monitor the effectiveness of risk controls.
- Improve Product Quality and Reliability: ISO 14971 integrates risk management techniques, such as FMEA and FTA, to prevent design and process failures, thereby enhancing the overall reliability of medical devices.
- Effective Benefit-Risk Analysis: ISO 14971 provides a structured method for evaluating and demonstrating that the benefits of a device outweigh its risks, supporting justifications for high-risk and innovative devices.
- Reduce Costs from Failures and Recalls: ISO 14971 identifies hazards that reduce the likelihood of post-market failures, recalls, and legal liabilities, thereby minimizing overall operational costs.
- Gain Competitive Market Advantage: ISO 14971 builds market trust and strengthens brand reputation by showcasing a transparent and robust approach to risk control and medical device safety assurance.
- Streamline Audits and Inspections: ISO 14971 maintains an up-to-date risk management file with documented risk assessment, control measures, and post-market surveillance data, facilitating efficient audits and inspections.
- Support Innovation with Safety: ISO 14971 facilitates the safe implementation of emerging technologies by providing a structured framework for identifying novel hazards, assessing associated risks, and implementing appropriate controls.
What Is the Structure of ISO 14971?
ISO 14971 is structured into ten (10) clauses and three (3) informative annexes.
The 10 clauses of ISO 14971 are listed below.
- Clause 1 Scope: Defines ISO 14971’s applicability to all stages of the medical device lifecycle, from design to post-market, for all device types and classifications.
- Clause 2 Normative References: States that ISO 14971 is self-contained, with no external normative references required for implementation.
- Clause 3 Terms and Definitions: Standardizes key risk-related terms (e.g., hazard, risk, residual risk) to ensure consistent understanding and application.
- Clause 4 General Requirements for Risk Management System: Outlines the framework, responsibilities, and documentation requirements for implementing a risk management system.
- Clause 5 Risk Analysis: Covers identification of hazards and hazardous situations, assessment of risk severity, and probability of occurrence estimation.
- Clause 6 Risk Evaluation: Evaluate risks against defined acceptability criteria to determine whether individual risks require mitigation, forming the basis for informed decision-making.
- Clause 7 Risk Control: Select and apply appropriate risk control measures, verify their implementation and effectiveness, and evaluate residual risks after mitigation.
- Clause 8 Evaluation of Overall Residual Risk: Perform a holistic review of all residual risks, and conduct benefit-risk analysis to justify the acceptability of medical devices where needed.
- Clause 9 Risk Management Review: Document a comprehensive review confirming that all risk-related activities are completed and the medical device is considered safe for use.
- Clause 10 Production and Post-Production Information: Mandates continuous data collection and feedback to manage risks in the post-market.
The three (3) annexes of ISO 14971 are listed below.
- Annex A Rationale for Requirements: Explain the intent and reasoning behind each clause to support a more profound understanding and proper application of the standard.
- Annex B Risk Management Process for Medical Devices: Provides an overview of the entire risk management process.
- Annex C Fundamental Risk Concepts: Define foundational risk concepts such as hazards, hazardous situations, harm, and probability of occurrence to strengthen interpretation and implementation consistency.
What Are the Key ISO 14971 Requirements?
The key ISO 14971 requirements for implementing a structured risk management process for medical devices are listed below.
- Clause 4 General Requirements for Risk Management System
- Clause 5 Risk Analysis
- Clause 6 Risk Evaluation
- Clause 7 Risk Control
- Clause 8 Evaluation of Overall Residual Risk
- Clause 9 Risk Management Review
- Clause 10 Production and Post-Production Activities
Clause 4 General Requirements for Risk Management System
Clause 4, general requirements for the risk management system, of ISO 14971 outlines the general requirements for a documented risk management system applicable to all medical device manufacturers, including those for software and in vitro diagnostic (IVD) devices. Clause 4 supports risk-related activities from design to post-market surveillance.
Manufacturers must develop a systematic process to identify, estimate, evaluate, and control risks, assign qualified personnel with defined responsibilities, and maintain evidence of competence. A structured risk management plan must define the scope and requirements of the risk management process. A device risk management file must include details of risk assessments and control measures for identified hazards and residual risk evaluation. Clause 4 establishes the framework and documentation to demonstrate controlled, auditable, transparent risk management as a continuous part of the quality system.
The main requirements of Clause 4 are listed below.
- Risk Management Process: Document and maintain a structured process to identify, assess, control, and monitor risks throughout the entire medical device lifecycle.
- Management Responsibilities: Designate qualified individuals to oversee risk management activities, establish risk acceptability criteria, and periodically review the suitability of the risk management process, ensuring top-level commitment to safety.
- Competence of Personnel: Review that all personnel involved in risk-related tasks possess the necessary education, training, and experience, and retain evidence of qualifications.
- Risk Management Plan: Establish a risk management plan defining scope, responsibilities, review requirements, risk acceptability criteria, and verification activities.
- Risk Management File: Compile and manage all risk-related records, including risk assessment, control measures, and residual risk evaluations, ensuring compliance with relevant standards, regulations, and stakeholder expectations.
Clause 5 Risk Analysis
Clause 5 risk analysis of ISO 14971 defines requirements to document the intended use of a medical device, identify hazards, and estimate risks by evaluating the probability of occurrence and severity of harm.
Clause 5 ensures that manufacturers conduct a complete and structured hazard identification and analysis process. Clause 5 supports consistent medical device risk assessment and provides the foundation for evaluating whether risks require control in the next steps of ISO 14971.
The main sections of clause 5 risk analysis are listed below.
- Risk Analysis Process: Conduct risk analysis and maintain a record in the risk management file, including a description of the medical device, scope, and responsible personnel.
- Intended Use and Reasonably Foreseeable Misuse: Document the intended use of the medical device and anticipate reasonably foreseeable misuse scenarios that may expose users to harm.
- Identification of Characteristics Related to Safety: Determine device characteristics (e.g., materials, energy sources, interface features) that may impact safety under normal or fault conditions.
- Identification of Hazards and Hazardous Situations: List potential sources of harm (hazards) and the circumstances (hazardous situations) under which they may lead to harm.
- Risk Estimation: Evaluate the probability of occurrence and severity of harm for each identified hazardous situation, forming the basis for risk evaluation in Clause 6.
Clause 6 Risk Evaluation
Clause 6 risk evaluation of ISO 14971 outlines the risk evaluation requirements used to determine whether the risks estimated from identified hazardous situations are acceptable based on pre-established risk acceptability criteria. Clause 6 bridges the outputs from Clause 5 risk analysis with the decision-making process that governs whether risk control measures are required.
Clause 6 ensures that risk acceptability decisions are made consistently and transparently, thereby reducing bias and promoting evidence-based medical device risk management.
For each hazardous situation, the manufacturer must do the following under clause 6.
- Evaluate the estimated risk using predefined acceptability criteria from the risk management plan.
- Treat it as a residual risk if the risk is acceptable, and proceed to Clause 7.6 (Completeness of Risk Control).
- Perform risk control activities as outlined in Clauses 7.1 to 7.6 if the risk is not acceptable.
- Document all results of the risk evaluation in the risk management file.
Clause 7 Risk Control
Clause 7 risk control of ISO 14971 defines requirements for selecting, implementing, and verifying control measures to reduce medical device risks to acceptable levels. Risk control applies to all risks deemed unacceptable during evaluation and ensures mitigation through documented, justified actions.
Clause 7 prioritizes risk controls in the following order: (1) inherent safety by design, (2) protective measures, and (3) safety information. Risk control measures must be verified for implementation and effectiveness. In clause 7, residual risk must be evaluated for acceptability or subjected to benefit-risk analysis.
Clause 7 enforces a disciplined, auditable, lifecycle-oriented process to ensure that mitigation is effective, traceable, and compliant with regulations.
The main sections of clause 7 risk control are listed below.
- Risk Control Option Analysis: Identify suitable control measures and apply them in order of priority: (1) inherently safe design and manufacturing, (2) protective measures in design or process, and (3) safety information and user training.
- Implementation of Risk Control Measures: Apply selected risk control measures and verify both their implementation and effectiveness. Document verification results, such as design validation or process qualification.
- Residual Risk Evaluation: After implementing controls, assess residual risk against the acceptability criteria defined in the risk management plan. Consider additional controls under 7.1 if risks remain unacceptable.
- Benefit-Risk Analysis: Perform a structured analysis to confirm that clinical or user benefits outweigh the risks when residual risks remain unacceptable and further reduction is impractical. Revise the device or intended use if still unacceptable.
- Risks Arising from Risk Control Measures: Evaluate potential adverse consequences, such as new hazards or increased risks, resulting from the implementation of controls. Manage any emerging risks using the same steps outlined in Clauses 5.5 through 7.4.
- Completeness of Risk Control: Perform a final review to ensure all identified hazardous situations are addressed and that no steps are omitted. Record findings in the risk management file for traceability and audit readiness.
Clause 8 Evaluation of Overall Residual Risk
Clause 8 defines requirements for evaluating overall residual risk by determining if the combined risks, after applying all control measures, are acceptable relative to the device’s intended medical benefit.
After implementing and verifying all risk control measures, the manufacturer must evaluate overall residual risk by assessing the combined impact of all individual residual risks against the benefits of the device’s intended use. This evaluation must follow the methodology and acceptability criteria outlined in the risk management plan.
The manufacturer must inform users of significant residual risks in the device documentation if the overall risk is acceptable. The manufacturer must implement additional controls or modify the device or its use if the overall risk is not acceptable. All evaluation results, justifications, decisions, and communications must be documented to ensure traceability, transparency, and compliance.
Clause 9 Risk Management Review
Clause 9 risk management review outlines the requirements related to verifying the completeness, adequacy, and proper execution of the entire risk management process before the medical device is released for commercial distribution.
Clause 9 ensures that a thorough review of the risk management process is conducted by qualified personnel at the end of the design and development phase. Clause 9 confirms that all planned risk management tasks have been completed, all risks have been evaluated, residual risk is deemed acceptable, and the device is safe for its intended use. The review also confirms that mechanisms are in place for ongoing production and post-production monitoring.
The results of the risk management review shall be recorded and maintained as the risk management report and shall be included in the risk management file.
Clause 10 Production and Post-Production Activities
Clause 10 of the production and post-production activities of ISO 14971 requires manufacturers to collect, review, and act on information from both the production and post-market phases of a medical device.
Clause 10 ensures that risk management continues after the device is released by monitoring real-world performance, identifying any new or emerging risks, and updating the risk documentation to confirm that existing safety measures remain effective.
Sections under clause 10 are listed below.
- General: Set up and maintain a documented process to actively collect, monitor, and analyze safety-related information during production and post-market use of the device.
- Information Collection: Monitor and collect data from production, process monitoring, users, service teams, suppliers, distribution channels, public databases, literature, incident reports, technologies, and competitor devices.
- Information Review: Evaluate if new hazards or hazardous situations have emerged, acceptable risks have become unacceptable, residual risk is no longer justified by benefits, or updated controls are needed due to changes in the state of the art.
- Actions: If new or changed risks are identified, take device-specific actions (e.g., reassessing the risk profile, modifying control measures, considering device updates, corrections, or recalls) or take process-level actions (e.g., evaluating implications for the overall risk management system, updating procedures, and feeding insights into management reviews).
How to Implement ISO 14971 Risk Management to Medical Devices?
To implement ISO 14971 risk management for medical devices effectively, the following steps should be followed, according to Annex B.2 of the ISO 14971 standard.
- Conduct Risk Analysis: Identify hazards and hazardous situations related to intended use and reasonably foreseeable misuse of the device; estimate the probability of occurrence and severity of potential harm associated with the hazardous situations identified.
- Perform Risk Evaluation: Determine whether the identified risks are acceptable based on predefined criteria or if further risk control actions are needed.
- Implement Risk Control Measures: Apply risk control measures, such as design changes, safety features, or enhancement of user information, to eliminate or reduce unacceptable risks to acceptable levels.
- Evaluate Overall Residual Risk: Assess the combined residual risks and decide whether they are acceptable or require a formal benefit-risk analysis. If deemed unacceptable, additional controls or device modification may be required.
- Review Risk Management Activities: Ensure all risk management steps are complete, documented, and aligned with the organization’s Quality Management System (QMS), as required by ISO 13485. Integration with the QMS is crucial because it embeds risk management into product lifecycle processes, enabling proactive safety measures and regulatory alignment.
- Monitor Post-Production Performance: Collect and analyze real-world data, including user feedback, complaints, and field reports, to identify emerging risks and update the risk management file as needed.
The image below provides an overview of risk management activities as applied to medical devices, as outlined in Annex B.2 of ISO 14971.
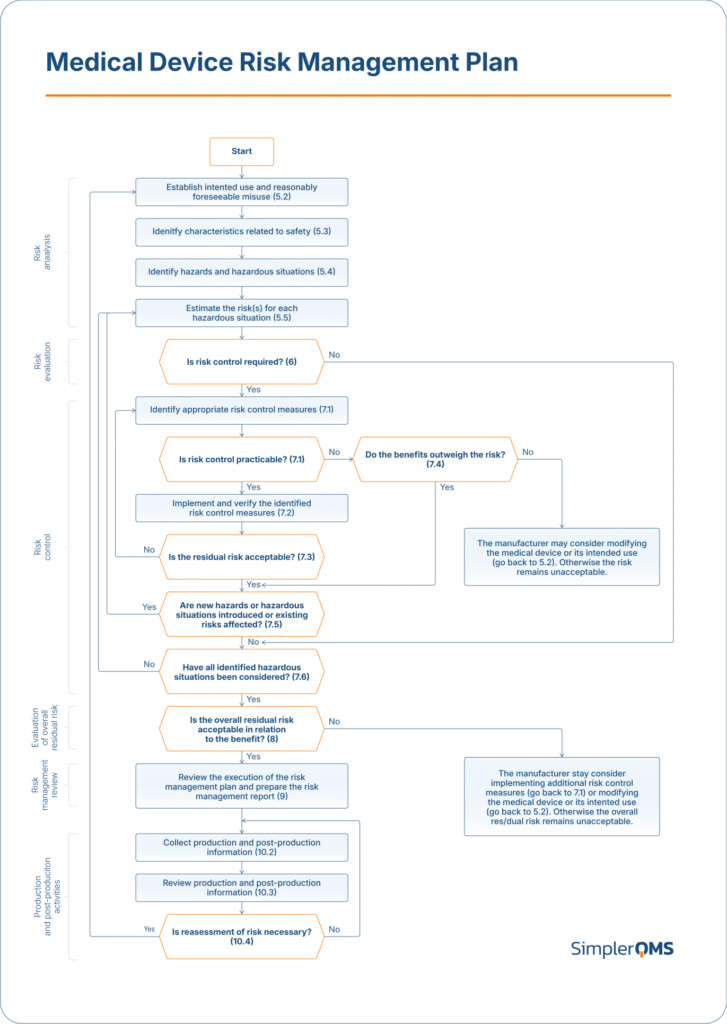
What Is the Difference Between ISO 14971 and ISO 13485?
The primary difference between ISO 14971 and ISO 13485 lies in their functional focus. ISO 14971 provides a framework for risk management throughout the lifecycle of a medical device, while ISO 13485 defines the requirements for a quality management system (QMS) specific to medical device manufacturers.
ISO 14971 provides a structured framework for risk management throughout the entire lifecycle of a medical device, guiding manufacturers in identifying, analyzing, evaluating, controlling, and continuously monitoring risks. ISO 14971 integrates directly with ISO 13485, serving as a risk-based foundation that supports and informs the quality system.
In contrast, ISO 13485 defines the QMS requirements for medical device manufacturers, ensuring consistency in design, development, production, and post-market surveillance. ISO 13485 emphasizes compliance with regulatory and customer expectations. Meanwhile, ISO 14971 ensures that all quality processes are highlighted by proactive risk assessment and mitigation, aligning safety with product performance.
What Is the Difference Between ISO 14971 and ICH Q9?
The main difference between ISO 14971 and ICH Q9 is their industry application and regulatory focus. While ISO 14971 and ICH Q9 are both risk management requirements, ISO 14971 applies to medical devices and ICH Q9 applies to pharmaceuticals.
ISO 14971 is the risk management standard for medical devices. ISO 14971 provides a systematic process for identifying, analyzing, evaluating, controlling, and monitoring risks associated with the safety and performance of medical devices, including the evaluation of residual risks and benefit-risk analysis.
On the other hand, ICH Q9 is the risk management guideline for pharmaceuticals, developed by the International Council for Harmonization (ICH). ICH Q9 outlines Quality Risk Management (QRM) principles used in the development, manufacturing, testing, and distribution of pharmaceutical drugs to ensure product quality and patient safety.
How Does Quality Management Software Support ISO 14971 Compliance?
Quality management software supports ISO 14971 compliance by digitizing, automating, and connecting essential risk management activities throughout the medical device lifecycle. This includes documenting risk analysis, implementing controls, evaluating residual risks, and monitoring post-market data.
SimplerQMS offers a medical device QMS software designed to help meet the requirements of both ISO 14971 and ISO 13485. SimplerQMS provides a cloud-based, GAMP 5 validated platform that integrates key QMS processes. These processes include risk management, document control, CAPA, design control, nonconformance, supplier management, audits, training, and others.
SimplerQMS supports ISO 14971 compliance through its risk management module, which offers the following capabilities.
- Centralized Risk Documentation: Consolidates risk analysis, risk assessments, and control measures into a single, traceable system integrated with all quality processes.
- Template-Driven and Configurable: Offers ready-to-use templates for risk assessments, plans, and traceability matrices, with drag-and-drop support for importing existing Word, Excel, or PDF formats.
- Automated Workflows: Guides users through compliant risk processes using built-in workflows, task notifications, and periodic risk review scheduling.
- Document Traceability and Linking: Enables bi-directional linking of risk files with SOPs, CAPAs, audits, suppliers, deviations, and other relevant documents, ensuring informed decision-making and audit readiness.
- Custom Fields and Filtering: Supports metadata customization (e.g., risk, methodology, severity level) and advanced search filters for personalized data insights.
- Audit and Regulatory Readiness: Facilitates fast document retrieval during audits and supports compliance with ISO 14971:2019, ISO 13485:2016, EU MDR, EU IVDR, FDA 21 CFR Part 11, and more.
- Traceability Matrix: Maintains end-to-end visibility by connecting documents, risks, mitigations, and regulatory chapters, enhancing risk control throughout the lifecycle.
- Integrated eQMS Environment: Operates within a full QMS platform covering document control, training, change control, CAPA, audit, complaint, and supplier management.