
The EU IVDR medical device classification under Regulation (EU) 2017/746 assigns devices into four risk classes: Class A, B, C, and D. The EU IVDR categorizes IVD medical devices based on their intended use and potential risk to individual and public health.
The EU IVDR device classification ensures that each device undergoes the appropriate level of regulatory scrutiny before entering the EU market.
Annex VIII of the EU IVDR outlines the classification rules that determine the risk class of IVD medical devices. Rules 1 to 7 assess the device’s diagnostic purpose, user type (professional or layperson), and clinical risk.
The correct classification under IVDR is essential because it determines the appropriate conformity assessment pathway, the level of Notified Body involvement, and the technical documentation and performance evaluations required.
Medical device companies increasingly rely on electronic quality management systems (eQMS) to meet the rigorous demands of EU IVDR.
SimplerQMS provides a GAMP5-validated eQMS platform built for medical device companies. SimplerQMS complies with FDA 21 CFR Part 11 and EU GMP Annex 11 requirements for electronic records and signatures. Additionally, SimplerQMS supports compliance with EU IVDR, EU MDR, ISO 13485, and 21 CFR Part 820 in the context of electronic quality management systems.
What are the EU IVDR Medical Device Classes?
According to Annex VIII of the EU IVDR, also known as Regulation (EU) 2017/746, IVD medical devices are categorized into four distinct categories: Class A, Class B, Class C, and Class D.
The class of an IVD medical device is determined by its intended purpose and the potential risks posed to individual and public health.
The EU IVDR medical device classes are listed below.
- Class A (Low Individual Risk, Low Public Health Risk): Class A includes devices with minimal risk to individuals and public health. Notified body involvement is not required, unless the device is sterile.
- Class B (Moderate Individual Risk, Low Public Health Risk): Class B devices carry moderate risk to the individual but limited public health implications. Class B devices require notified body conformity assessment (includes technical documentation assessment and review of performance evaluation).
- Class C (High Individual Risk, Moderate Public Health Risk): Devices in Class C present a high individual risk and moderate public health concern. Class C devices require notified body conformity assessment (includes technical documentation assessment, review of performance evaluation, and summary of safety and performance).
- Class D (High Individual Risk, High Public Health Risk): Class D covers the highest-risk devices, which have critical implications for patient safety and public health. This device class undergoes the most stringent notified body conformity assessment, including batch testing and third-party laboratory verification.
The EU IVDR device classes (A–D) against key regulatory aspects are shown in the table below.
| Aspect | Class A | Class B | Class C | Class D |
|---|---|---|---|---|
| Risk Level | Low individual risk, low public health risk | Moderate individual risk, low public health risk | High individual risk, moderate public health risk | High individual risk, high public health risk |
| Conformity Assessment | Self-certification (except sterile) | Notified Body required | Notified Body required | Notified Body + EU Reference Laboratory (EURL) |
| Sterility Review | This applies if the device is sterile | This applies if the device is sterile | This applies if the device is sterile | This applies if the device is sterile |
| Approval Route | Annex IV Annex IX or XI (sterility) | Annex IX | Annex IX or X + XI | Annex IX + EURL or X + XI + EURL |
| EU Reference Laboratory | Not required | Not required | Not required | Required for some high-risk devices |
| Examples | Specimen containers, laboratory equipment | Pregnancy tests, urinalysis strips | Cancer diagnostics, companion diagnostics | HIV tests, ABO/Rh blood typing, Hepatitis B and Hepatitis C tests |
What Is EU IVDR Class A Medical Device?
EU IVDR Class A medical devices are categorized as in vitro diagnostic (IVD) devices with low individual risk and low public health risk, as defined in Annex VIII of Regulation (EU) 2017/746. Class A represents the lowest risk level under the IVDR framework.
Class A devices are typically used in supportive roles in diagnostics rather than generating diagnostic results, and the consequences of malfunction are minimal. Manufacturers are permitted to self-certify non-sterile Class A devices without the involvement of a Notified Body. However, for sterile Class A devices, a Notified Body must assess the aspects related to sterility as part of the conformity assessment process.
What Are EU IVDR Class A Medical Device Examples?
Several examples of Class A Devices under EU IVDR are listed below.
- Specimen Receptacles: Collect and transport biological specimens, intended solely for sample handling.
- Buffer Solutions: Used to preserve or stabilize samples, which are chemical reagents essential for maintaining sample integrity.
- General Laboratory Instruments: Support general laboratory procedures with laboratory instruments such as beakers, centrifuge tubes, and other basic tools.
- Histological Stains: Enhance contrast in tissue samples through histological stains, without contributing to diagnostic interpretation.
- Non-Sterile Sample Containers: Utilized for basic storage of biological specimens.
- Pipettes and Pipette Tips: Transfer precise liquid volumes in test preparation with pipettes and pipette tips.
- Instruments used for IVD procedures: Examples include enzyme immunoassay analyzers, instruments for automated purification of nucleic acids
The examples of EU IVDR Class A Medical Devices are shown in the image below.

What Is EU IVDR Class B Medical Device?
EU IVDR Class B medical devices are IVD devices that present a moderate individual risk and a low public health risk. Class B devices provide diagnostic information but are not typically used for life-threatening conditions or high-risk treatment decisions.
Class B devices differ from Class A devices in terms of risk level and clinical significance. Class A devices pose a low risk to individual and public health and typically serve auxiliary roles such as sample collection or laboratory support. In contrast, Class B devices present a moderate individual risk and generate diagnostic information that informs routine clinical decisions. While manufacturers may self-certify non-sterile Class A devices, Class B devices require certification by a Notified Body.
What Are EU IVDR Class B Medical Device Examples?
The common examples of EU IVDR Class B medical devices are listed below.
- Pregnancy Self-Tests: Detect early pregnancy through hormone (hCG) detection.
- Ovulation Prediction Tests: Identify fertile windows by detecting luteinizing hormone surges.
- CRP (C-reactive protein) Tests: Assess inflammation markers that may indicate infection or chronic disease status.
- Urinalysis Strips: Test urine for glucose or for urinary tract infection in point-of-care environments.
- Cholesterol-Detecting Devices: Monitor cholesterol levels, such as high-density lipoprotein (HDL) and low-density lipoprotein (LDL).
The examples of EU IVDR Class B Medical Devices are shown in the image below.

What Is EU IVDR Class C Medical Device?
EU IVDR Class C medical devices are IVD devices that present a high individual risk and a moderate public health risk. Class C devices are commonly used to detect serious health conditions such as cancer, sexually transmitted infections (STIs), or genetic disorders that require significant clinical management.
Class C devices exert a greater influence on diagnostic and treatment decisions than Class B devices, which are typically used for routine monitoring or diagnosis of non-critical conditions. Both Class B and Class C devices require Notified Body involvement, but Class C devices are subject to more rigorous scrutiny due to their potential clinical impact.
What Are EU IVDR Class C Medical Device Examples?
Several EU IVDR Class C medical device examples are listed below.
- Immune Status in Prenatal Screening (e.g., CMV tests for pregnant women): Helps assess fetal risk and maternal immunity.
- Companion Diagnostics: Support personalized medicine by guiding eligibility for specific therapies, such as EGFR for lung cancer, BRAF for melanoma, and KRAS for colorectal cancer.
- Cancer Screening, Diagnosis, or Staging: Detection and determining cancer progression, such as PAP tests and CIN tests
- Genetic Tests in Humans: Includes carrier screening (e.g., BRCA1, BRCA2 genes for cancer), disease predisposition (e.g., HLA-DQ2/DQ8 for celiac disease), and pharmacogenomic markers.
- Genetic Disorders in Embryos or Fetuses: Test for prenatal conditions such as Non-Invasive Prenatal Testing (NIPT) for trisomy or microdeletions.
The EU IVDR Class C Medical Devices are summarized in the image below.

What Is EU IVDR Class D Medical Device?
EU IVDR Class D medical devices represent the highest risk category, posing a high individual and a high public health risk. Class D IVD devices are used for screening, diagnosing, or monitoring life-threatening and highly transmissible infectious diseases.
Given Class D devices’ critical public health role, any diagnostic error could lead to widespread consequences. Compared to Class C devices, which address serious but less contagious conditions, Class D devices undergo the most stringent regulatory oversight.
Class D devices include Notified Body certification and mandatory assessment by an EU Reference Laboratory before market placement.
What Are EU IVDR Class D Medical Device Examples?
The examples of EU IVDR Class D medical devices are listed below.
- Devices for Transmissible Pathogens: Detect highly pathogenic and epidemic/pandemic-prone agents such as the Ebola virus, HIV, and multi-resistant gram-negative bacteria (MRGN).
- Monitor Degree of Infection in Life-Threatening Conditions: Used for ongoing disease management and to ensure the proper course of treatment in serious infections (e.g., Hepatitis B Virus, Hepatitis C Virus, hemorrhagic fever viruses)
- Assess Donor Suitability: Used to assess donor suitability for blood, tissue, and organ donation to prevent transmission of infectious disease (e.g., Screening for HIV, Hepatitis B, and Hepatitis C).
- Determine Blood Grouping and Immuno-Compatibility: Determine transfusion medicine and transplant compatibility (e.g., ABO and Rhesus (Rh) typing, antigen systems like Kell, Kidd, and Duffy).
The EU IVDR Class D Medical Devices are summarized in the image below.
What Are EU IVDR Classification Rules?
The EU IVDR classification rules, as defined in Annex VIII, are listed below.
- Rule 1: Rule 1 applies to devices that detect transmissible agents in blood, tissues, cells, or organs to assess donor suitability, devices used to detect life-threatening highly transmissible diseases, and those used to determine infectious load where critical for patient management. Examples include HIV, HBV, HCV, and Ebola. Due to their potential for widespread health impact, if misdiagnosed, these devices are classified as Class D.
- Rule 2: Rule 2 governs IVDs used for blood grouping and tissue typing, including tests for ABO, Rh, Kell, Duffy, and Kidd markers. Given the potentially fatal risk of misclassification during transfusions or transplantations, these are designated as Class D.
- Rule 3: Rule 3 covers IVDs intended to diagnose or monitor life-threatening or serious diseases that do not pose a high public health concern, such as infectious disease, cancer, genetic disorders, and congenital conditions. Devices under this rule, including BRCA gene tests, cancer markers, and companion diagnostics, are typically classified as Class C.
- Rule 4: Rule 4 pertains to self-testing devices that laypersons use. Depending on the condition detected, these devices may fall under Class C (e.g., blood glucose) or Class B (e.g., cholesterol, pregnancy tests). Rule 4 also addresses near-patient testing performed outside centralized laboratories by professionals. Near-patient tests are classified in their own right, depending on the intended purpose and associated risk. Common examples include rapid HIV tests (Class D), cardiac marker monitoring (Class C), and rapid strep tests (Class B)
- Rule 5: Rule 5 applies to general laboratory devices that support IVD testing without producing results. Instruments specifically used for IVD procedures and specimen receptacles. Rule 5 includes general-purpose lab instruments such as pipettes, buffer solutions, and specimen containers as well as IVD instruments such as enzyme immunoassay analyzers. These are typically Class A, reflecting their lower inherent risk. However, if an instrument has an independent measuring function, it is classified according to the intended purpose of the analysis (e.g., cell counting analyzers).
- Rule 6: Rule 6 is a catch-all for devices not covered by the previous rules. These devices influence clinical decisions but do not pose a high public health or individual risk and as such are classified as Class B.
- Rule 7: Rule 7 covers IVD devices which are controls without any assigned qualitative or quantitative value for their analytes. These controls are classified as Class B (eg unassigned control sera or QC material).
Rule 1
Rule 1 classifies IVD medical devices as Class D if they are intended for the scenario listed below.
- Detect transmissible agents in blood, cells, tissues, or organs to assess donor suitability for transfusion, transplantation, or cell administration.
- Detect life-threatening infectious diseases with a high or suspected high risk of transmission.
- Determine the infectious load in life-threatening conditions, where ongoing monitoring is critical for effective patient management.
Rule 1 covers devices with the highest combined individual and public health risk. These medical devices are essential for ensuring the safety of blood and tissue products, controlling epidemic-prone diseases, and guiding treatment in serious infectious conditions. Inaccurate or missed results could lead to outbreaks, unsafe donations, or treatment failures, posing serious threats to both patients and the public.
Devices falling under Rule 1 are mandatory and classified as Class D due to their direct involvement in protecting recipients and the general population from serious health threats.
Rule 2
Rule 2 applies to IVD devices used for blood grouping or tissue typing to determine immunological compatibility during transfusions, transplantations, or cell therapies. Devices under this rule are classified as Class C, by default. However, if the device is intended to detect high-risk blood cell markers, it is reclassified to Class D.
Rule 2 addresses the serious risks of immunological incompatibility, such as acute hemolytic reactions, which can occur if high-risk markers are mismatched. Because these reactions may be life-threatening, devices that detect clinically critical markers are placed under Class D to ensure maximum regulatory oversight.
Devices that detect lower-risk markers remain in Class C, reflecting their comparatively reduced clinical impact.
Rule 3
Rule 3 applies to IVD medical devices used for the following.
- Detecting infectious diseases not listed under Rule 1.
- Pre-natal screening to determine immune status towards transmissible agents
- Diagnosing cancer, congenital disorders, genetic predispositions, disease staging, or patient management where there is significant risk associated with erroneous results.
- Companion diagnostics
- Devices under Rule 3 are typically classified as Class C, reflecting their significant role in clinical decision-making for serious conditions, that do not pose a high public health concern.
Rule 3 acknowledges that while the conditions diagnosed under Rule 3 may not carry immediate public health threats, they pose high individual risk and often guide complex or long-term treatment strategies. The diagnostic output significantly impacts clinical interventions, such as surgery, chemotherapy, or genetic counseling. Therefore, higher regulatory control (Class C) is required to ensure accuracy, reliability, and safety.
Rule 4
Rule 4 applies to IVD devices for self-testing or near-patient testing.
- Rule 4a covers IVD devices intended for self-testing by non-professional users (laypersons). These devices are designed for use outside clinical settings, typically at home, without supervision by healthcare professionals. A default classification of Class C is applied to self-testing devices (e.g., blood glucose meters, blood clotting)
- However, if the test poses a lower health risk, the device is classified as Class B (e.g., pregnancy, cholesterol)
Rule 4a reflects the risks associated with unsupervised testing, including misinterpretation of results, delayed treatment, or psychological impact. Since these devices are used by individuals without medical training, the accuracy, clarity of instructions, and reliability of results are critical. Higher classification ensures additional safeguards depending on the condition’s risk level.
Rule 4b applies to IVD devices intended for near-patient testing, meaning they are used outside of central laboratories, typically in point-of-care settings by trained professionals (e.g., general practitioners, emergency rooms, field clinics). These devices deliver rapid results and may be used to support immediate clinical decisions.
All near-patient tests are classified in their own right, they can be Class D, C, or B depending on the intended purpose and associated risk.
This classification of devices for near-patient testing accounts for the context in which tests are used, particularly where rapid decisions depend on immediate results. Although healthcare professionals operate these tests, the lack of centralized laboratory procedures heightens dependence on the test’s reliability.
Rule 4 classification ensures devices used outside controlled laboratory environments are subject to appropriate regulatory scrutiny, reflecting the higher risk of misuse, misinterpretation, or harm due to incorrect results.
Rule 5
Rule 5a applies to general laboratory devices that support laboratory processes without directly generating diagnostic results. General-purpose tools and reagents used for in vitro diagnostic examinations fall under this category. Rule 5b applies to instruments that are specifically used for IVD procedures and Rule 5c covers receptacles used for storing biological specimens for IVD tests.
Although these devices do not directly pose diagnostic risks, their role in testing can affect the reliability or safety of diagnostic procedures. They are typically classified as Class A, reflecting their lower inherent risk. However, if an instrument has an independent measuring function, it is classified according to the intended purpose of the analysis (e.g., cell counting analyzers).
The rationale for Rule 5 is to ensure that all components involved in IVD testing, even those without a direct diagnostic function, are subject to appropriate regulatory oversight, particularly where their design or function could affect the accuracy or safety of testing procedures.
Rule 6
Rule 6 applies to devices not covered under rules 1-5. These devices influence clinical decisions but do not pose a high public health or individual risk, and as such, they are classified as Class B.
Rule 7
Rule 7 covers IVD devices that are controlled without any assigned qualitative or quantitative value for their analytes. These controls are classified as Class B (e.g., unassigned control sera or QC material).
What is the Difference Between EU IVDR and EU MDR Medical Device Classification?
The main difference between EU IVDR and EU MDR medical device classification lies in the type of products regulated and the risk criteria applied.
EU IVDR governs IVD medical devices, which examine human specimens such as blood, urine, or tissue to provide diagnostic information. Based on their potential risk to individual and public health, devices are classified into Class A, B, C, or D under EU IVDR.
In contrast, EU MDR applies to all other medical devices, including implants, surgical instruments, and medical software used for diagnosing, preventing, or monitoring disease. EU MDR devices are categorized into Class I, IIa, IIb, or III, depending on their invasiveness, duration of body contact, and overall risk.
While EU IVDR prioritizes diagnostic reliability and safeguards for public health, EU MDR concentrates on the safety, performance, and clinical risks associated with therapeutic and interventional medical devices.
What is the Difference Between EU IVDR and FDA Medical Device Classification?
The main distinction between EU IVDR and FDA medical device classification lies in their area of regulatory framework application and risk-based categorization criteria.
EU IVDR specifically classifies IVD medical devices into Class A, B, C, or D. The classification is based on factors such as intended purpose, individual risk, and public health impact. Devices in Class D represent the highest risk, requiring Notified Body assessment and EU Reference Laboratory testing before market placement. EU IVDR compliance is essential for placing an IVD on the EU market.
On the other hand, the FDA classifies all medical devices, including IVDs, into Class I, II, or III based primarily on the risk posed to patients and how much control is needed to keep them safe and effective according to 21 CFR 860 Medical Device Classification Procedures. Class I devices are low-risk and often exempt from premarket submission. Class II devices require a 510(k) premarket notification. Class III poses the most significant risk and requires rigorous Premarket Approval (PMA). FDA compliance is mandatory for any medical device intended for distribution within the United States.
What are the Different EU IVDR Medical Device Regulatory Approval Pathways?
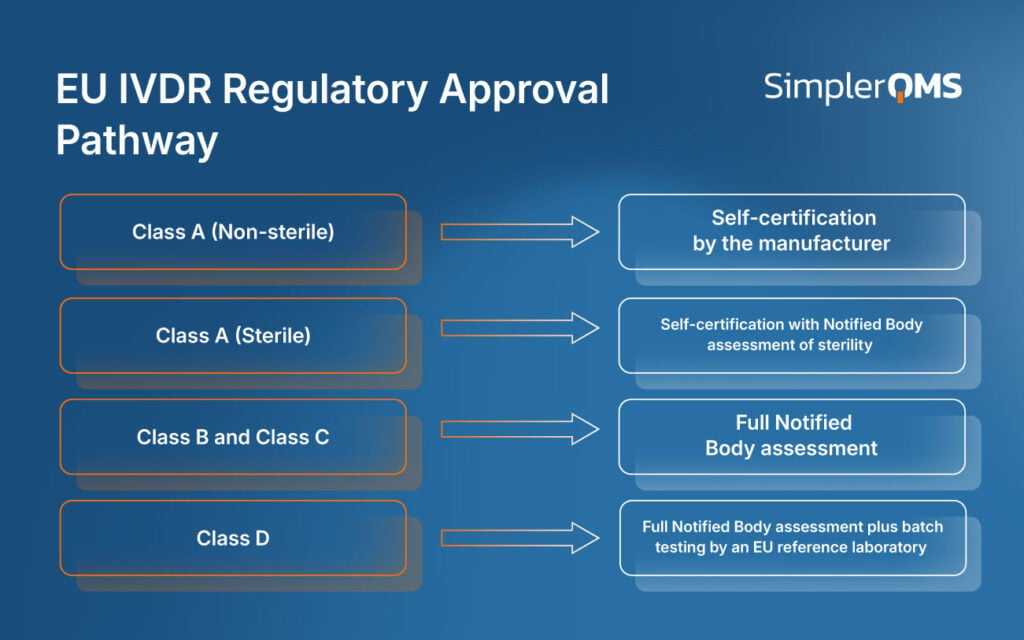
The different regulatory approval pathways for EU IVDR medical devices depend on the device classification, intended use, and associated risk.
Each class follows a defined conformity assessment route, determining the level of regulatory review and Notified Body involvement. Higher-risk classifications correspond to more complex approval processes.
Class A non-sterile devices may undergo self-certification. Class A (sterile), B, C, and D devices require Notified Body review, with Class D additionally assessed by EU Reference Laboratories. This risk-based model ensures that devices with greater potential impact on public and patient health receive stricter evaluation before market entry.
The different pathways for EU IVDR medical devices are illustrated below.
EU IVDR Class A Medical Device Approval Pathway
The EU IVDR Class A medical device approval pathway represents the least burdensome regulatory route, allowing manufacturers to self-certify for non-sterile devices.
To comply, manufacturers must implement a Quality Management System (QMS) and prepare technical documentation in accordance with Annexes II and III. Other requirements include performing a self-declared conformity assessment under Annex IV, assigning a Unique Device Identifier (UDI), and registering the device in EUDAMED. Additionally, robust post-market surveillance (PMS) and vigilance systems must be maintained.
For sterile Class A devices, a Notified Body (Annex IX or Annex XI) is involved only to assess the aspects of the manufacturing process related to sterility. At the same time, all other conformity elements remain under the manufacturer’s responsibility.
EU IVDR Class B Medical Device Approval Pathway
The EU IVDR Class B medical device approval pathway requires a full conformity assessment by a Notified Body before a device can be placed on the EU market.
Manufacturers must comply with the requirements below.
- Implement a compliant QMS following Annex IX.
- Compile complete technical documentation under Annex II and III.
- Undergo Notified Body review of documentation, including performance evaluation.
- Assign a UDI and register the device and related data in EUDAMED.
- Establish and maintain PMS and vigilance procedures.
EU IVDR Class C Medical Device Approval Pathway
The EU IVDR Class C medical device approval pathway involves a comprehensive conformity assessment conducted by a Notified Body, reflecting the higher individual risk associated with these devices.
Manufacturers must comply with the requirements below.
- Apply QMS and choose a conformity assessment route. Annex IX: Full QMS with product technical documentation review or Annex X combined with Annex XI: Type examination with production quality assurance.
- Compile technical documentation under Annex II and III, including a performance evaluation (scientific validity, analytical, and clinical performance) and summary of safety and performance.
- Undergo Notified Body review of the documentation and compliance with GSPRs.
- Assign a UDI and register the device in EUDAMED.
- Maintain PMS, PSUR, and vigilance systems.
EU IVDR Class D Medical Device Approval Pathway
The EU IVDR Class D medical device approval pathway involves the most rigorous conformity assessment, requiring oversight by both the Notified Body and the EURL. EURL involvement is required where an EU Reference Laboratory has been designated for that device type under Commission Implementing Regulation (EU) 2022/944 of June 17, 2022.
Manufacturers are required to meet the following requirements.
- Apply a full QMS and follow one of the following routes: Annex IX: Full QMS with product documentation, or Annex X combined with Annex XI: Type examination plus production quality assurance.
- Submit detailed technical documentation under Annex II and III, including performance evaluation reports covering scientific validity, analytical, and clinical performance of the device, and a summary of safety and performance.
- Undergo Notified Body assessment and batch testing for verification of performance claims by an EU Reference Laboratory (where designated or equivalent).
- Assign a UDI and register both the device and the manufacturer in EUDAMED.
- Implement PMS, including vigilance and submission of PSUR.
What is the Role of eQMS in Facilitating EU IVDR CE Marking Regulatory Process?
An electronic Quality Management System (eQMS) is a digital platform designed to manage, automate, and centralize quality processes, documentation, and compliance activities related to medical devices.
Within the framework of EU IVDR, an eQMS plays a critical role in achieving and maintaining CE marking by ensuring that all documentation, traceability, and performance evidence meet regulatory requirements. Electronic QMS supports manufacturers in structuring and maintaining an audit-ready quality system. The system helps streamline the preparation and submission of technical documentation, performance evaluation reports, and other compliance deliverables. With these functions, an eQMS helps ensure regulatory alignment, reduce manual errors, and accelerate conformity assessments under IVDR.
The main capabilities of the medical device eQMS are listed below.
- Document Control: Centralizes and provides version control for technical documentation, design control, SOPs, and classification evidence.
- Training Management: Automates training and competency tracking to ensure personnel are competent to do the job.
- Audit and CAPA Management: Enables traceable internal audits and CAPA workflows aligned with ISO 13485 and IVDR conformity assessments.
- Supplier and Change Management: Tracks supplier qualification and change controls tied to classification-specific performance and regulatory documents.
- Risk Management: Integrates risk assessments into centralized documentation systems, ensuring accessibility during audits, conformity assessments, or Notified Body reviews.
- Product Lifecycle Management: Connects technical documents and performance evaluation reports in the product lifecycle into a centralized system to support EU IVDR compliance.
Implementing an eQMS offers significant advantages for organizations seeking EU IVDR compliance. QMS software enhances efficiency by streamlining the management of regulatory deliverables, including technical files, performance evaluations, and PMS documentation. Automated workflows reduce human error, ensuring critical processes such as CAPA, document control, and change management are consistently executed. Real-time traceability and centralized data access enhance audit readiness. Additionally, an eQMS supports scalability, making it easier to manage increasing documentation volume and team expansion as the company grows.
SimplerQMS provides fully validated QMS software according to GAMP5. SimplerQMS complies with FDA 21 CFR Part 11 and EU Annex 11 requirements for using electronic records, electronic signatures, and computerized systems in regulated environments.
SimplerQMS helps medical device companies ensure compliance with applicable QMS requirements, including EU IVDR, EU MDR, ISO 13485, and others. The system offers comprehensive QMS process support, including document control, change control, design control, training, CAPA, audits, supplier management, and vigilance reporting.